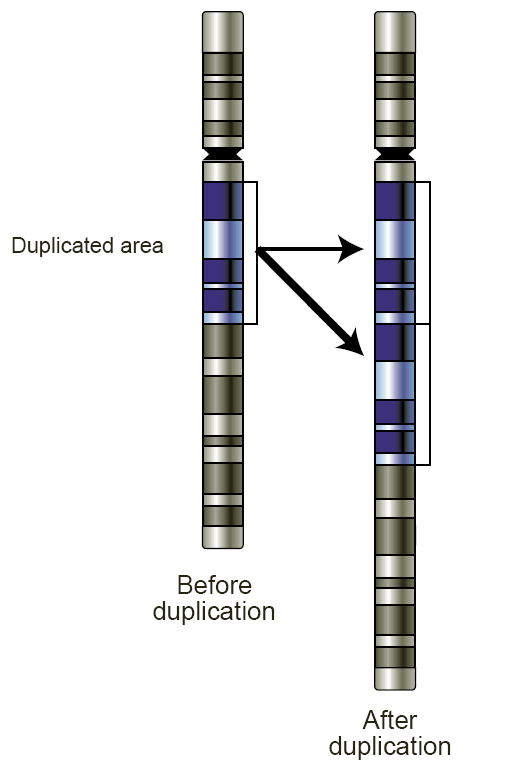
Copy number variation
Copy number variation (CNV) is a phenomenon in which sections of the genome are repeated and the number of repeats in the genome varies between individuals.[1] Copy number variation is a type of structural variation: specifically, it is a type of duplication or deletion event that affects a considerable number of base pairs.[2] Approximately two-thirds of the entire human genome may be composed of repeats[3] and 4.8–9.5% of the human genome can be classified as copy number variations.[4] In mammals, copy number variations play an important role in generating necessary variation in the population as well as disease phenotype.[1]
Copy number variations can be generally categorized into two main groups: short repeats and long repeats. However, there are no clear boundaries between the two groups and the classification depends on the nature of the loci of interest. Short repeats include mainly dinucleotide repeats (two repeating nucleotides e.g. A-C-A-C-A-C...) and trinucleotide repeats. Long repeats include repeats of entire genes. This classification based on size of the repeat is the most obvious type of classification as size is an important factor in examining the types of mechanisms that most likely gave rise to the repeats,[5] hence the likely effects of these repeats on phenotype.
Types and chromosomal rearrangements[edit]
One of the most well known examples of a short copy number variation is the trinucleotide repeat of the CAG base pairs in the huntingtin gene responsible for the neurological disorder Huntington's disease.[6] For this particular case, once the CAG trinucleotide repeats more than 36 times in a trinucleotide repeat expansion, Huntington's disease will likely develop in the individual and it will likely be inherited by his or her offspring.[6] The number of repeats of the CAG trinucleotide is inversely correlated with the age of onset of Huntington's disease.[7] These types of short repeats are often thought to be due to errors in polymerase activity during replication including polymerase slippage, template switching, and fork switching which will be discussed in detail later. The short repeat size of these copy number variations lends itself to errors in the polymerase as these repeated regions are prone to misrecognition by the polymerase and replicated regions may be replicated again, leading to extra copies of the repeat.[8] In addition, if these trinucleotide repeats are in the same reading frame in the coding portion of a gene, it may lead to a long chain of the same amino acid, possibly creating protein aggregates in the cell,[7] and if these short repeats fall into the non-coding portion of the gene, it may affect gene expression and regulation. On the other hand, a variable number of repeats of entire genes is less commonly identified in the genome. One example of a whole gene repeat is the alpha-amylase 1 gene (AMY1) that encodes alpha-amylase which has a significant copy number variation between different populations with different diets.[9] Although the specific mechanism that allows the AMY1 gene to increase or decrease its copy number is still a topic of debate, some hypotheses suggest that the non-homologous end joining or the microhomology-mediated end joining is likely responsible for these whole gene repeats.[9] Repeats of entire genes has immediate effects on expression of that particular gene, and the fact that the copy number variation of the AMY1 gene has been related to diet is a remarkable example of recent human evolutionary adaptation.[9] Although these are the general groups that copy number variations are grouped into, the exact number of base pairs copy number variations affect depends on the specific loci of interest. Currently, using data from all reported copy number variations, the mean size of copy number variant is around 118kb, and the median is around 18kb.[10]
In terms of the structural architecture of copy number variations, research has suggested and defined hotspot regions in the genome where copy number variations are four times more enriched.[2] These hotspot regions were defined to be regions containing long repeats that are 90–100% similar known as segmental duplications either tandem or interspersed and most importantly, these hotspot regions have an increased rate of chromosomal rearrangement.[2] It was thought that these large-scale chromosomal rearrangements give rise to normal variation and genetic diseases, including copy number variations.[1] Moreover, these copy number variation hotspots are consistent throughout many populations from different continents, implying that these hotspots were either independently acquired by all the populations and passed on through generations, or they were acquired in early human evolution before the populations split, the latter seems more likely.[1] Lastly, spatial biases of the location at which copy number variations are most densely distributed does not seem to occur in the genome.[1] Although it was originally detected by fluorescent in situ hybridization and microsatellite analysis that copy number repeats are localized to regions that are highly repetitive such as telomeres, centromeres, and heterochromatin,[11] recent genome-wide studies have concluded otherwise.[2] Namely, the subtelomeric regions and pericentromeric regions are where most chromosomal rearrangement hotspots are found, and there is no considerable increase in copy number variations in that region.[2] Furthermore, these regions of chromosomal rearrangement hotspots do not have decreased gene numbers, again, implying that there is minimal spatial bias of the genomic location of copy number variations.[2]
Detection and identification[edit]
Copy number variation was initially thought to occupy an extremely small and negligible portion of the genome through cytogenetic observations.[12] Copy number variations were generally associated only with small tandem repeats or specific genetic disorders,[13] therefore, copy number variations were initially only examined in terms of specific loci. However, technological developments led to an increasing number of highly accurate ways of identifying and studying copy number variations. Copy number variations were originally studied by cytogenetic techniques, which are techniques that allow one to observe the physical structure of the chromosome.[12] One of these techniques is fluorescent in situ hybridization (FISH) which involves inserting fluorescent probes that require a high degree of complementarity in the genome for binding.[10] Comparative genomic hybridization was also commonly used to detect copy number variations by fluorophore visualization and then comparing the length of the chromosomes.[10]
Recent advances in genomics technologies gave rise to many important methods that are of extremely high genomic resolution and as a result, an increasing number of copy number variations in the genome have been reported.[10] Initially these advances involved using bacterial artificial chromosome (BAC) array with around 1 megabase of intervals throughout the entire gene,[14] BACs can also detect copy number variations in rearrangement hotspots allowing for the detection of 119 novel copy number variations.[2] High throughput genomic sequencing has revolutionized the field of human genomics and in silico studies have been performed to detect copy number variations in the genome.[2] Reference sequences have been compared to other sequences of interest using fosmids by strictly controlling the fosmid clones to be 40kb.[15] Sequencing end reads would provide adequate information to align the reference sequence to the sequence of interest, and any misalignments are easily noticeable thus concluded to be copy number variations within that region of the clone.[15] This type of detection technique offers a high genomic resolution and precise location of the repeat in the genome, and it can also detect other types of structural variation such as inversions.[10]
In addition, another way of detecting copy number variation is using single nucleotide polymorphisms (SNPs).[10] Due to the abundance of the human SNP data, the direction of detecting copy number variation has changed to utilize these SNPs.[16] Relying on the fact that human recombination is relatively rare and that many recombination events occur in specific regions of the genome known as recombination hotspots, linkage disequilibrium can be used to identify copy number variations.[16] Efforts have been made in associating copy number variations with specific haplotype SNPs by analyzing the linkage disequilibrium, using these associations, one is able to recognize copy number variations in the genome using SNPs as markers. Next-generation sequencing techniques including short and long read sequencing are nowadays increasingly used and have begun to replace array-based techniques to detect copy number variations.[17][18]
Brain cells[edit]
Among the neurons in the human brain, somatically derived copy number variations are frequent.[28] Copy number variations show wide variability (9 to 100% of brain neurons in different studies). Most alterations are between 2 and 10 Mb in size with deletions far outnumbering amplifications.[28]
Genomic duplication and triplication of the gene appear to be a rare cause of Parkinson's disease, although more common than point mutations.[29]
Copy number variants in RCL1 gene are associated with a range of neuropsychiatric phenotypes in children.[30]