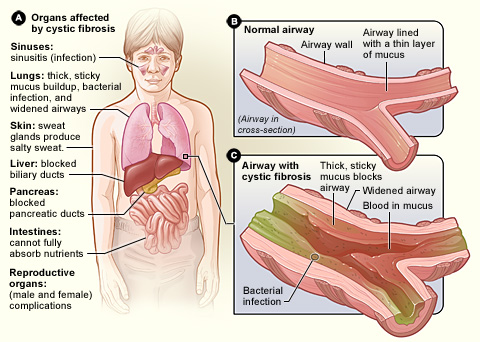
Cystic fibrosis
Cystic fibrosis (CF) is a genetic disorder inherited in an autosomal recessive manner that impairs the normal clearance of mucus from the lungs, which facilitates the colonization and infection of the lungs by bacteria, notably Staphylococcus aureus.[6] CF is a rare[7][8] genetic disorder that affects mostly the lungs, but also the pancreas, liver, kidneys, and intestine.[1][9] The hallmark feature of CF is the accumulation of thick mucus in different organs. Long-term issues include difficulty breathing and coughing up mucus as a result of frequent lung infections.[1] Other signs and symptoms may include sinus infections, poor growth, fatty stool, clubbing of the fingers and toes, and infertility in most males.[1] Different people may have different degrees of symptoms.[1]
Cystic fibrosis
Mucoviscidosis
Difficulty breathing, coughing up mucus, poor growth, fatty stool[1]
Symptoms recognizable ~6 month[2]
Long term[3]
Genetic (autosomal recessive)[1]
Genetic
Life expectancy between 42 and 50 years (developed world)[5]
1 out of 3,000 (Northern European)[1]
Cystic fibrosis is inherited in an autosomal recessive manner.[1] It is caused by the presence of mutations in both copies (alleles) of the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) protein.[1] Those with a single working copy are carriers and otherwise mostly healthy.[3] CFTR is involved in the production of sweat, digestive fluids, and mucus.[10] When the CFTR is not functional, secretions that are usually thin instead become thick.[11] The condition is diagnosed by a sweat test and genetic testing.[1] The sweat test measures sodium concentration, as people with cystic fibrosis have abnormally salty sweat, which can often be tasted by parents kissing their children. Screening of infants at birth takes place in some areas of the world.[1]
There is no known cure for cystic fibrosis.[3] Lung infections are treated with antibiotics which may be given intravenously, inhaled, or by mouth.[1] Sometimes, the antibiotic azithromycin is used long-term.[1] Inhaled hypertonic saline and salbutamol may also be useful.[1] Lung transplantation may be an option if lung function continues to worsen.[1] Pancreatic enzyme replacement and fat-soluble vitamin supplementation are important, especially in the young.[1] Airway clearance techniques such as chest physiotherapy may have some short-term benefit, but long-term effects are unclear.[12] The average life expectancy is between 42 and 50 years in the developed world,[5][13] with a median of 40.7 years.[14] Lung problems are responsible for death in 70% of people with cystic fibrosis.[1]
CF is most common among people of Northern European ancestry, for whom it affects about 1 out of 3,000 newborns,[1] and among which around 1 out of 25 people is a carrier.[3] It is least common in Africans and Asians, though it does occur in all races.[1] It was first recognized as a specific disease by Dorothy Andersen in 1938, with descriptions that fit the condition occurring at least as far back as 1595.[9] The name "cystic fibrosis" refers to the characteristic fibrosis and cysts that form within the pancreas.[9][15]