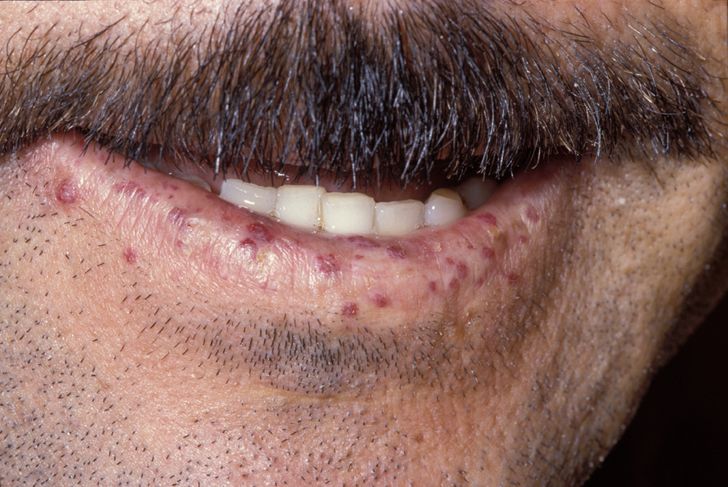
Hereditary hemorrhagic telangiectasia
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler–Weber–Rendu disease and Osler–Weber–Rendu syndrome, is a rare autosomal dominant genetic disorder that leads to abnormal blood vessel formation in the skin, mucous membranes, and often in organs such as the lungs, liver, and brain.[1][2]
Hereditary Hemorrhagic Telangiectasia
It may lead to nosebleeds, acute and chronic digestive tract bleeding, and various problems due to the involvement of other organs. Treatment focuses on reducing bleeding from blood vessel lesions, and sometimes surgery or other targeted interventions to remove arteriovenous malformations in organs. Chronic bleeding often requires iron supplements and sometimes blood transfusions. HHT is transmitted in an autosomal dominant fashion, and occurs in one in 5,000–8,000 people in North America.[1][2]
The disease carries the names of Sir William Osler, Henri Jules Louis Marie Rendu, and Frederick Parkes Weber, who described it in the late 19th and early 20th centuries.[3]
History[edit]
Several 19th century English physicians, starting with Henry Gawen Sutton (1836–1891)[24] and followed by Benjamin Guy Babington (1794–1866)[25] and John Wickham Legg (1843–1921),[26] described the most common features of HHT, particularly the recurrent nosebleeds and the hereditary nature of the disease. The French physician Henri Jules Louis Marie Rendu (1844–1902) observed the skin and mucosal lesions, and distinguished the condition from hemophilia.[27] The Canadian-born Sir William Osler (1849–1919), then at Johns Hopkins Hospital and later at Oxford University, made further contributions with a 1901 report in which he described characteristic lesions in the digestive tract.[28] The English physician Frederick Parkes Weber (1863–1962) reported further on the condition in 1907 with a series of cases.[29] The term "hereditary hemorrhagic telangiectasia" was first used by the American physician Frederic M. Hanes (1883–1946) in a 1909 article on the condition.[3][30]
The diagnosis of HHT remained a clinical one until the genetic defects that cause HHT were identified by a research group at Duke University Medical Center, in 1994 and 1996 respectively.[9][10] In 2000, the international scientific advisory committee of cureHHT formerly called the HHT Foundation International published the now widely used Curaçao criteria.[7][15] In 2006, a group of international experts met in Canada and formulated an evidence-based guideline, sponsored by cureHHT.[7] This guideline has since been updated in 2020 and can be found here.