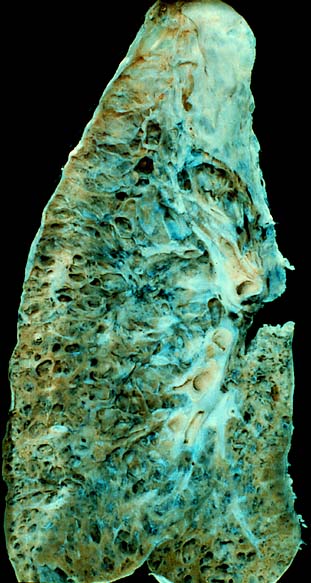
Pulmonary fibrosis
Pulmonary fibrosis is a condition in which the lungs become scarred over time.[1] Symptoms include shortness of breath, a dry cough, feeling tired, weight loss, and nail clubbing.[1] Complications may include pulmonary hypertension, respiratory failure, pneumothorax, and lung cancer.[2]
Pulmonary fibrosis
Interstitial pulmonary fibrosis
Shortness of breath, dry cough, feeling tired, weight loss, nail clubbing[1]
Tobacco smoking, environmental pollution, certain medications, connective tissue diseases, interstitial lung disease, unknown[1][3]
Poor[3]
>5 million people[5]
Causes include environmental pollution, certain medications, connective tissue diseases, infections, and interstitial lung diseases.[1][3][6] However, in most cases the cause is unknown, and termed idiopathic pulmonary fibrosis.[1][3] Diagnosis may be based on symptoms, medical imaging, lung biopsy, and lung function tests.[1]
No cure exists and only limited treatment options are available.[1] Treatment is directed towards efforts to improve symptoms and may include oxygen therapy and pulmonary rehabilitation.[1][4] Certain medications may be used to try to slow the worsening of scarring.[4] Lung transplantation may occasionally be an option.[3] At least 5 million people are affected globally.[5] Life expectancy is generally less than five years.[3]
Symptoms of pulmonary fibrosis are mainly:[1]
Pulmonary fibrosis is suggested by a history of progressive shortness of breath (dyspnea) with exertion. Sometimes fine inspiratory crackles can be heard at the lung bases on auscultation. A chest X-ray may or may not be abnormal, but high-resolution CT will frequently demonstrate abnormalities.[3]
Pulmonary fibrosis may be a secondary effect of other diseases. Most of these are classified as interstitial lung diseases. Examples include autoimmune disorders, viral infections and bacterial infection like tuberculosis which may cause fibrotic changes in both lung's upper or lower lobes and other microscopic injuries to the lung. However, pulmonary fibrosis can also appear without any known cause. In this case, it is termed "idiopathic".[7] Most idiopathic cases are diagnosed as idiopathic pulmonary fibrosis. This is a diagnosis of exclusion of a characteristic set of histologic/pathologic features known as usual interstitial pneumonia (UIP). In either case, there is a growing body of evidence which points to a genetic predisposition in a subset of patients. For example, a mutation in surfactant protein C (SP-C) has been found to exist in some families with a history of pulmonary fibrosis.[8] Autosomal dominant mutations in the TERC or TERT genes, which encode telomerase, have been identified in about 15 percent of pulmonary fibrosis patients.[9]
Diseases and conditions that may cause pulmonary fibrosis as a secondary effect include:[3][8]
Pulmonary fibrosis involves a gradual replacement of normal lung tissue with fibrotic tissue. Such scar tissue causes an irreversible decrease in oxygen diffusion capacity, and the resulting stiffness or decreased compliance makes pulmonary fibrosis a restrictive lung disease.[14]
Pulmonary fibrosis is perpetuated by aberrant wound healing, rather than chronic inflammation.[15]
It is the main cause of restrictive lung disease that is intrinsic to the lung parenchyma. In contrast, quadriplegia[16] and kyphosis[17] are examples of causes of restrictive lung disease that do not necessarily involve pulmonary fibrosis.
Common genes implicated in fibrosis are Transforming Growth Factor-Beta (TGF-β),[18] Connective Tissue Growth Factor (CTGF),[19] Epidermal Growth Factor Receptor (EGFR),[20] Interleukin-13 (IL-13),[21] Platelet-Derived Growth Factor (PDGF),[22] Wnt/β-catenin signaling pathway[23] and TNIK.[24]
Pulmonary fibrosis creates scar tissue. The scarring is permanent once it has developed.[28] Slowing the progression and prevention depends on the underlying cause:
The immune system is felt to play a central role in the development of many forms of pulmonary fibrosis. The goal of treatment with immune suppressive agents such as corticosteroids is to decrease lung inflammation and subsequent scarring. Responses to treatment are variable. Those whose conditions improve with immune suppressive treatment probably do not have idiopathic pulmonary fibrosis, for idiopathic pulmonary fibrosis has no significant treatment or cure. [29]
Prognosis[edit]
Hypoxia caused by pulmonary fibrosis can lead to pulmonary hypertension, which, in turn, can lead to heart failure of the right ventricle. Hypoxia can be prevented with oxygen supplementation.[3]
Pulmonary fibrosis may also result in an increased risk for pulmonary emboli, which can be prevented by anticoagulants.[3]