p53
p53, also known as Tumor protein P53, cellular tumor antigen p53 (UniProt name), or transformation-related protein 53 (TRP53) is a regulatory protein that is often mutated in human cancers. The p53 proteins (originally thought to be, and often spoken of as, a single protein) are crucial in vertebrates, where they prevent cancer formation.[5] As such, p53 has been described as "the guardian of the genome" because of its role in conserving stability by preventing genome mutation.[6] Hence TP53[note 1] is classified as a tumor suppressor gene.[7][8][9][10][11]
For other uses, see P53 (disambiguation).The TP53 gene is the most frequently mutated gene (>50%) in human cancer, indicating that the TP53 gene plays a crucial role in preventing cancer formation.[5] TP53 gene encodes proteins that bind to DNA and regulate gene expression to prevent mutations of the genome.[12] In addition to the full-length protein, the human TP53 gene encodes at least 12 protein isoforms.[13]
Function[edit]
DNA damage and repair[edit]
p53 plays a role in regulation or progression through the cell cycle, apoptosis, and genomic stability by means of several mechanisms:
Regulation[edit]
p53 acts as a cellular stress sensor. It is normally kept at low levels by being constantly marked for degradation by the E3 ubiquitin ligase protein MDM2.[43] p53 is activated in response to myriad stressors – including DNA damage (induced by either UV, IR, or chemical agents such as hydrogen peroxide), oxidative stress,[44] osmotic shock, ribonucleotide depletion, viral lung infections[45] and deregulated oncogene expression. This activation is marked by two major events. First, the half-life of the p53 protein is increased drastically, leading to a quick accumulation of p53 in stressed cells. Second, a conformational change forces p53 to be activated as a transcription regulator in these cells. The critical event leading to the activation of p53 is the phosphorylation of its N-terminal domain. The N-terminal transcriptional activation domain contains a large number of phosphorylation sites and can be considered as the primary target for protein kinases transducing stress signals.
The protein kinases that are known to target this transcriptional activation domain of p53 can be roughly divided into two groups. A first group of protein kinases belongs to the MAPK family (JNK1-3, ERK1-2, p38 MAPK), which is known to respond to several types of stress, such as membrane damage, oxidative stress, osmotic shock, heat shock, etc. A second group of protein kinases (ATR, ATM, CHK1 and CHK2, DNA-PK, CAK, TP53RK) is implicated in the genome integrity checkpoint, a molecular cascade that detects and responds to several forms of DNA damage caused by genotoxic stress. Oncogenes also stimulate p53 activation, mediated by the protein p14ARF.
In unstressed cells, p53 levels are kept low through a continuous degradation of p53. A protein called Mdm2 (also called HDM2 in humans), binds to p53, preventing its action and transports it from the nucleus to the cytosol. Mdm2 also acts as an ubiquitin ligase and covalently attaches ubiquitin to p53 and thus marks p53 for degradation by the proteasome. However, ubiquitylation of p53 is reversible. On activation of p53, Mdm2 is also activated, setting up a feedback loop. p53 levels can show oscillations (or repeated pulses) in response to certain stresses, and these pulses can be important in determining whether the cells survive the stress, or die.[46]
MI-63 binds to MDM2, reactivating p53 in situations where p53's function has become inhibited.[47]
A ubiquitin specific protease, USP7 (or HAUSP), can cleave ubiquitin off p53, thereby protecting it from proteasome-dependent degradation via the ubiquitin ligase pathway. This is one means by which p53 is stabilized in response to oncogenic insults. USP42 has also been shown to deubiquitinate p53 and may be required for the ability of p53 to respond to stress.[48]
Recent research has shown that HAUSP is mainly localized in the nucleus, though a fraction of it can be found in the cytoplasm and mitochondria. Overexpression of HAUSP results in p53 stabilization. However, depletion of HAUSP does not result in a decrease in p53 levels but rather increases p53 levels due to the fact that HAUSP binds and deubiquitinates Mdm2. It has been shown that HAUSP is a better binding partner to Mdm2 than p53 in unstressed cells.
USP10, however, has been shown to be located in the cytoplasm in unstressed cells and deubiquitinates cytoplasmic p53, reversing Mdm2 ubiquitination. Following DNA damage, USP10 translocates to the nucleus and contributes to p53 stability. Also USP10 does not interact with Mdm2.[49]
Phosphorylation of the N-terminal end of p53 by the above-mentioned protein kinases disrupts Mdm2-binding. Other proteins, such as Pin1, are then recruited to p53 and induce a conformational change in p53, which prevents Mdm2-binding even more. Phosphorylation also allows for binding of transcriptional coactivators, like p300 and PCAF, which then acetylate the C-terminal end of p53, exposing the DNA binding domain of p53, allowing it to activate or repress specific genes. Deacetylase enzymes, such as Sirt1 and Sirt7, can deacetylate p53, leading to an inhibition of apoptosis.[50] Some oncogenes can also stimulate the transcription of proteins that bind to MDM2 and inhibit its activity.
Experimental analysis of p53 mutations[edit]
Most p53 mutations are detected by DNA sequencing. However, it is known that single missense mutations can have a large spectrum from rather mild to very severe functional effects.[59]
The large spectrum of cancer phenotypes due to mutations in the TP53 gene is also supported by the fact that different isoforms of p53 proteins have different cellular mechanisms for prevention against cancer. Mutations in TP53 can give rise to different isoforms, preventing their overall functionality in different cellular mechanisms and thereby extending the cancer phenotype from mild to severe. Recent studies show that p53 isoforms are differentially expressed in different human tissues, and the loss-of-function or gain-of-function mutations within the isoforms can cause tissue-specific cancer or provide cancer stem cell potential in different tissues.[11][66][67][68] TP53 mutation also hits energy metabolism and increases glycolysis in breast cancer cells.[69]
The dynamics of p53 proteins, along with its antagonist Mdm2, indicate that the levels of p53, in units of concentration, oscillate as a function of time. This "damped" oscillation is both clinically documented [70] and mathematically modelled.[71][72] Mathematical models also indicate that the p53 concentration oscillates much faster once teratogens, such as double-stranded breaks (DSB) or UV radiation, are introduced to the system. This supports and models the current understanding of p53 dynamics, where DNA damage induces p53 activation (see p53 regulation for more information). Current models can also be useful for modelling the mutations in p53 isoforms and their effects on p53 oscillation, thereby promoting de novo tissue-specific pharmacological drug discovery.
Discovery[edit]
p53 was identified in 1979 by Lionel Crawford, David P. Lane, Arnold Levine, and Lloyd Old, working at Imperial Cancer Research Fund (UK) Princeton University/UMDNJ (Cancer Institute of New Jersey), and Memorial Sloan Kettering Cancer Center, respectively. It had been hypothesized to exist before as the target of the SV40 virus, a strain that induced development of tumors. The name p53 was given in 1979 describing the apparent molecular mass.
The TP53 gene from the mouse was first cloned by Peter Chumakov of The Academy of Sciences of the USSR in 1982,[73] and independently in 1983 by Moshe Oren in collaboration with David Givol (Weizmann Institute of Science).[74][75] The human TP53 gene was cloned in 1984[7] and the full length clone in 1985.[76]
It was initially presumed to be an oncogene due to the use of mutated cDNA following purification of tumor cell mRNA. Its role as a tumor suppressor gene was revealed in 1989 by Bert Vogelstein at the Johns Hopkins School of Medicine and Arnold Levine at Princeton University.[77][78] p53 went on to be identified as a transcription factor by Guillermina Lozano working at MD Anderson Cancer Center.[79]
Warren Maltzman, of the Waksman Institute of Rutgers University first demonstrated that TP53 was responsive to DNA damage in the form of ultraviolet radiation.[80] In a series of publications in 1991–92, Michael Kastan of Johns Hopkins University, reported that TP53 was a critical part of a signal transduction pathway that helped cells respond to DNA damage.[81]
In 1993, p53 was voted molecule of the year by Science magazine.[82]
Isoforms[edit]
As with 95% of human genes, TP53 encodes more than one protein. All these p53 proteins are called the p53 isoforms.[5] These proteins range in size from 3.5 to 43.7 kDa. Several isoforms were discovered in 2005, and so far 12 human p53 isoforms have been identified (p53α, p53β, p53γ, ∆40p53α, ∆40p53β, ∆40p53γ, ∆133p53α, ∆133p53β, ∆133p53γ, ∆160p53α, ∆160p53β, ∆160p53γ). Furthermore, p53 isoforms are expressed in a tissue dependent manner and p53α is never expressed alone.[11]
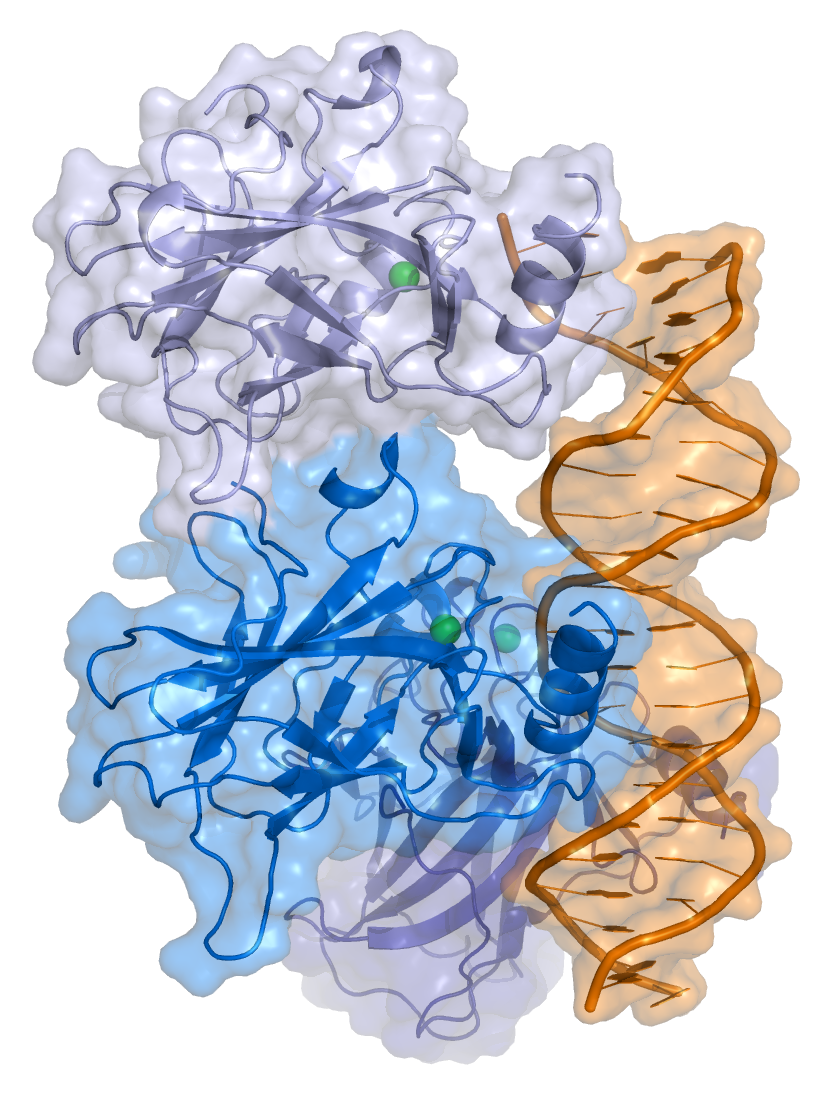
The full length p53 isoform proteins can be subdivided into different protein domains. Starting from the N-terminus, there are first the amino-terminal transcription-activation domains (TAD 1, TAD 2), which are needed to induce a subset of p53 target genes. This domain is followed by the proline rich domain (PXXP), whereby the motif PXXP is repeated (P is a proline and X can be any amino acid). It is required among others for p53 mediated apoptosis.[88] Some isoforms lack the proline rich domain, such as Δ133p53β,γ and Δ160p53α,β,γ; hence some isoforms of p53 are not mediating apoptosis, emphasizing the diversifying roles of the TP53 gene.[66] Afterwards there is the DNA binding domain (DBD), which enables the proteins to sequence specific binding. The C-terminus domain completes the protein. It includes the nuclear localization signal (NLS), the nuclear export signal (NES) and the oligomerisation domain (OD). The NLS and NES are responsible for the subcellular regulation of p53. Through the OD, p53 can form a tetramer and then bind to DNA. Among the isoforms, some domains can be missing, but all of them share most of the highly conserved DNA-binding domain.
The isoforms are formed by different mechanisms. The beta and the gamma isoforms are generated by multiple splicing of intron 9, which leads to a different C-terminus. Furthermore, the usage of an internal promoter in intron 4 causes the ∆133 and ∆160 isoforms, which lack the TAD domain and a part of the DBD. Moreover, alternative initiation of translation at codon 40 or 160 bear the ∆40p53 and ∆160p53 isoforms.[11]
Due to the isoformic nature of p53 proteins, there have been several sources of evidence showing that mutations within the TP53 gene giving rise to mutated isoforms are causative agents of various cancer phenotypes, from mild to severe, due to single mutation in the TP53 gene (refer to section Experimental analysis of p53 mutations for more details).