
Stevens–Johnson syndrome
Stevens–Johnson syndrome (SJS) is a type of severe skin reaction.[1] Together with toxic epidermal necrolysis (TEN) and Stevens–Johnson/toxic epidermal necrolysis (SJS/TEN) overlap, they are considered febrile mucocutaneous drug reactions and probably part of the same spectrum of disease, with SJS being less severe.[1][5][3] Erythema multiforme (EM) is generally considered a separate condition.[6] Early symptoms of SJS include fever and flu-like symptoms.[1] A few days later, the skin begins to blister and peel, forming painful raw areas.[1] Mucous membranes, such as the mouth, are also typically involved.[1] Complications include dehydration, sepsis, pneumonia and multiple organ failure.[1]
Not to be confused with Dubin–Johnson syndrome.Stevens–Johnson syndrome
Fever, skin blisters, skin peeling, painful skin, red eyes[1]
Age < 30[2]
Certain medications, certain infections, unknown[2][1]
<10% of the skin involved, skin biopsy[2]
Hospitalization, stopping the cause[2]
1–2 per million per year (together with TEN)[1]
The most common cause is certain medications such as lamotrigine, carbamazepine, allopurinol, sulfonamide antibiotics and nevirapine.[1] Other causes can include infections such as Mycoplasma pneumoniae and cytomegalovirus, or the cause may remain unknown.[2][1] Risk factors include HIV/AIDS and systemic lupus erythematosus.[1]
The diagnosis of Stevens–Johnson syndrome is based on involvement of less than 10% of the skin.[2] It is known as TEN when more than 30% of the skin is involved and considered an intermediate form when 10–30% is involved.[3] SJS/TEN reactions are believed to follow a type IV hypersensitivity mechanism.[7] It is also included with drug reaction with eosinophilia and systemic symptoms (DRESS syndrome), acute generalized exanthematous pustulosis (AGEP) and toxic epidermal necrolysis in a group of conditions known severe cutaneous adverse reactions (SCARs).[8]
Treatment typically takes place in hospital such as in a burn unit or intensive care unit.[2] Efforts may include stopping the cause, pain medication, antihistamines, antibiotics, intravenous immunoglobulins or corticosteroids.[2] Together with TEN, SJS affects 1 to 2 people per million per year.[1] Typical onset is under the age of 30.[2] Skin usually regrows over two to three weeks; however, complete recovery can take months.[2] Overall, the risk of death with SJS is 5 to 10%.[1][4]
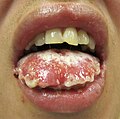
Inflammation and peeling of the lips—with sores presenting on the tongue and the mucous membranes in SJS

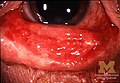
SJS usually begins with fever, sore throat, and fatigue, which is commonly misdiagnosed and therefore treated with antibiotics. SJS, SJS/TEN, and TEN are often heralded by fever, sore throat, cough, and burning eyes for 1 to 3 days.[9] Patients with these disorders frequently experience burning pain of their skin at the start of disease.[9] Ulcers and other lesions begin to appear in the mucous membranes, almost always in the mouth and lips, but also in the genital and anal regions. Those in the mouth are usually extremely painful and reduce the patient's ability to eat or drink. Conjunctivitis occurs in about 30% of children who develop SJS.[10] A rash of round lesions about an inch across arises on the face, trunk, arms and legs, and soles of the feet, but usually not the scalp.[11]
Prevention[edit]
Screening individuals for certain predisposing gene variants before initiating treatment with particular SJS-, TEN/SJS-, or TEN-inducing drugs is recommended or under study. These recommendations are typically limited to specific populations that show a significant chance of having the indicated gene variant since screening of populations with extremely low incidences of expressing the variant is considered cost-ineffective.[52] Individuals expressing the HLA allele associated with sensitivity to an indicated drug should not be treated with the drug. These recommendations include the following.[8][53] Before treatment with carbamazepine, the Taiwan and USA Food and Drug Administrations recommend screening for HLA-B*15:02 in certain Asian groups. This has been implemented in Taiwan, Hong Kong, Singapore, and many medical centers in Thailand and Mainland China. Before treatment with allopurinol, the American College of Rheumatology guidelines for managing gout recommend HLA-B*58:01 screening. This is provided in many medical centers in Taiwan, Hong Kong, Thailand, and Mainland China. Before treatment with abacavir, the USA Food and Drug Administration recommends screening for HLA-B*57:01 in Caucasian populations. This screening is widely implemented. It has also been suggested that all individuals found to express this HLA serotype avoid treatment with abacovir. Current trials are underway in Taiwan to define the cost-effectiveness of avoiding phenytoin in SJS, SJS/TEN, and TEN for individuals expressing the CYP2C9*3 allele of CYP2C9.[53]
Treatment[edit]
SJS constitutes a dermatological emergency. Patients with documented Mycoplasma infections can be treated with oral macrolide or oral doxycycline.[11]
Initially, treatment is similar to that for patients with thermal burns, and continued care can only be supportive (e.g., intravenous fluids and nasogastric or parenteral feeding) and symptomatic (e.g., analgesic mouth rinse for mouth ulcer). Dermatologists and surgeons tend to disagree about whether the skin should be debrided.[11]
Beyond this kind of supportive care, no treatment for SJS is accepted. Treatment with corticosteroids is controversial. Early retrospective studies suggested corticosteroids increased hospital stays and complication rates. No randomized trials of corticosteroids have been conducted for SJS, and it can be managed successfully without them.[11]
Other agents have been used, including cyclophosphamide and ciclosporin, but none have exhibited much therapeutic success. Intravenous immunoglobulin treatment has shown some promise in reducing the length of the reaction and improving symptoms. Other common supportive measures include the use of topical pain anesthetics and antiseptics, maintaining a warm environment, and intravenous analgesics.
An ophthalmologist should be consulted immediately, as SJS frequently causes the formation of scar tissue inside the eyelids, leading to corneal vascularization, impaired vision, and a host of other ocular problems. Those with chronic ocular surface disease caused by SJS may find some improvement with PROSE treatment (prosthetic replacement of the ocular surface ecosystem treatment).[54]
Prognosis[edit]
SJS (with less than 10% of body surface area involved) has a mortality rate of around 5%. The mortality for toxic epidermal necrolysis (TEN) is 30–40%. The risk for death can be estimated using the SCORTEN scale, which takes a number of prognostic indicators into account.[55] It is helpful to calculate a SCORTEN within the first 3 days of hospitalization.[9] Other outcomes include organ damage/failure, ocular morbidity, and blindness.[56][57] Restrictive lung disease may develop in patients with SJS and TEN after initial acute pulmonary involvement.[9] Patients with SJS or TEN caused by a drug have a better prognosis the earlier the causative drug is withdrawn.[9]
History[edit]
SJS is named for Albert Mason Stevens and Frank Chambliss Johnson, American pediatricians who jointly published a description of the disorder in the American Journal of Diseases of Children in 1922.[58][59]
Research[edit]
In 2015, the NIH and the Food and Drug Administration (FDA) organized a workshop entitled "Research Directions in Genetically-Mediated Stevens–Johnson Syndrome/Toxic Epidermal Necrolysis".[9]