Osteogenesis imperfecta
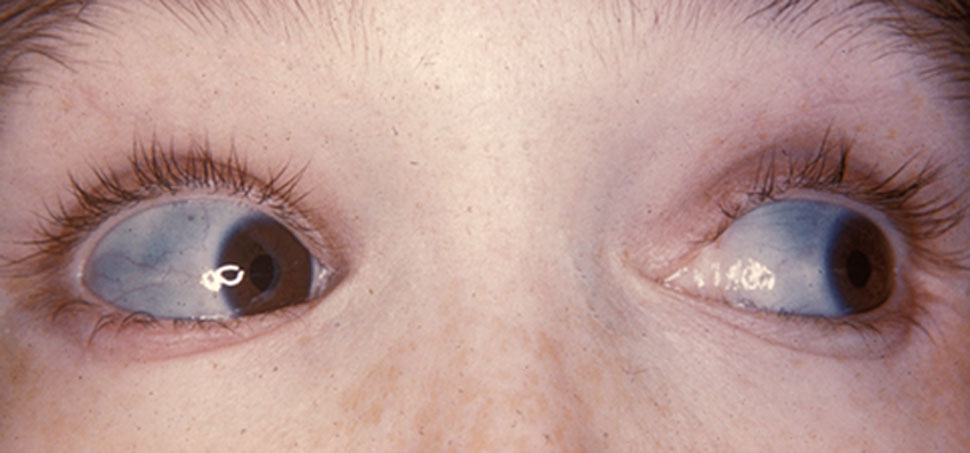
Osteogenesis imperfecta (IPA: /ˌɒstioʊˈdʒɛnəsɪs ˌɪmpɜːrˈfɛktə/;[4] OI), colloquially known as brittle bone disease, is a group of genetic disorders that all result in bones that break easily.[1]: 85 [9] The range of symptoms—on the skeleton as well as on the body's other organs—may be mild to severe.[5]: 1512 Symptoms found in various types of OI include whites of the eye (sclerae) that are blue instead, short stature, loose joints, hearing loss, breathing problems[10] and problems with the teeth (dentinogenesis imperfecta).[5] Potentially life-threatening complications, all of which become more common in more severe OI, include: tearing (dissection) of the major arteries, such as the aorta;[1]: 333 [11] pulmonary valve insufficiency secondary to distortion of the ribcage;[1]: 335–341 [12] and basilar invagination.[13]: 106–107
"Brittle bone disease" redirects here. For the etiologically distinct disease arising primarily from malnutrition rather than exclusively from genetic mutation, see rickets.Osteogenesis imperfecta (OI)
Bones that break easily, blue tinge to the sclera (whites of the eye), short height, joint hypermobility, hearing loss[5]
Birth
Long term
Based on symptoms, DNA testing
Healthy lifestyle (exercise, no smoking), metal rods through the long bones
Depends on the type
1 in 15,000–20,000 people[8]
The underlying mechanism is usually a problem with connective tissue due to a lack of, or poorly formed, type I collagen.[5]: 1513 In more than 90% of cases, OI occurs due to mutations in the COL1A1 or COL1A2 genes.[14] These mutations may be hereditary in an autosomal dominant manner but may also occur spontaneously (de novo).[9][15] There are four clinically defined types: type I, the least severe; type IV, moderately severe; type III, severe and progressively deforming; and type II, perinatally lethal.[9] As of September 2021, 19 different genes are known to cause the 21 documented genetically defined types of OI, many of which are extremely rare and have only been documented in a few individuals.[16][17] Diagnosis is often based on symptoms and may be confirmed by collagen biopsy or DNA sequencing.[10]
Although there is no cure,[10] most cases of OI do not have a major effect on life expectancy,[1]: 461 [15] death during childhood from it is rare,[10] and many adults with OI can achieve a significant degree of autonomy despite disability.[18] Maintaining a healthy lifestyle by exercising, eating a balanced diet sufficient in vitamin D and calcium, and avoiding smoking can help prevent fractures.[19] Genetic counseling may be sought by those with OI to prevent their children from inheriting the disorder from them.[1]: 101 Treatment may include acute care of broken bones, pain medication, physical therapy, mobility aids such as leg braces and wheelchairs,[10] vitamin D supplementation, and, especially in childhood, rodding surgery.[20] Rodding is an implantation of metal intramedullary rods along the long bones (such as the femur) in an attempt to strengthen them.[10] Medical research also supports the use of medications of the bisphosphonate class, such as pamidronate, to increase bone density.[21] Bisphosphonates are especially effective in children;[22] however, it is unclear if they either increase quality of life or decrease the rate of fracture incidence.[7]
OI affects only about one in 15,000 to 20,000 people, making it a rare genetic disease.[8] Outcomes depend on the genetic cause of the disorder (its type). Type I (the least severe) is the most common, with other types comprising a minority of cases.[15][23][24] Moderate-to-severe OI primarily affects mobility; if rodding surgery is performed during childhood, some of those with more severe types of OI may gain the ability to walk.[25] The condition has been described since ancient history.[26] The Latinate term osteogenesis imperfecta was coined by Dutch anatomist Willem Vrolik in 1849; translated literally, it means "imperfect bone formation".[26][27]: 683
Pathophysiology[edit]
People with OI are either born with defective connective tissue, born without the ability to make it in sufficient quantities, or, in the rarest genetic types, born with deficiencies in other aspects of bone formation such as chaperone proteins, the Wnt signaling pathway, the BRIL protein, et cetera.[16] In type I the collagen's structure itself is normal, it is just its quantity that is low.[5]: 1516 Types II, III and IV are usually, but not always, related to a deficiency of type I collagen.[93] One possible deficiency arises from an amino acid substitution of glycine to a bulkier amino acid, such as alanine, in the collagen protein's triple helix structure. The larger amino acid side-chains lead to steric effects that creates a bulge in the collagen complex, which in turn influences both the molecular nanomechanics and the interaction between molecules, which are both compromised.[94][95] Depending on both the location of the substitution and the amino acid being used instead, different effects are seen which account for the type diversity in OI despite the same two collagen genes being responsible for most cases.[96][95] Replacements of glycine with serine or cysteine are seen less often in fatal type II OI, while replacements with valine, aspartic acid, glutamic acid, or arginine are seen more often.[95]
At a larger scale, the relationship between the collagen fibrils and hydroxyapatite crystals to form bone is altered, causing brittleness.[96] Bone fractures occur because the stress state within collagen fibrils is altered at the locations of mutations, where locally larger shear forces lead to rapid failure of fibrils even at moderate loads because the homogeneous stress state normally found in healthy collagen fibrils is lost.[94] OI is therefore a multi-scale phenomenon, where defects at the smallest levels of tissues (genetic, nano, micro) domino to affect the macro level of tissues.[94]
Epidemiology[edit]
In the United States, the incidence of osteogenesis imperfecta is estimated to be one per 20,000 live births.[138] An estimated 20,000 to 50,000 people are affected by OI in the United States.[139]
The most common types are I, II, III, and IV, while the rest are very rare.[140] Type I is the most common and has been reported to be around three times more common than type II. The prevalence of types III and IV is less certain.[23] In a 1989 study in Denmark, type I was found to comprise 71% of cases and type II 12% of cases, with other types comprising the other 17%.[15] In a 2015 study in Sweden, type I was nearly six times more common than type III and nearly four times more common than type IV.[24]
Most people with OI receive it from a parent, but in many cases, it is a brand new (de novo or "sporadic") mutation in a family. Among a study of patients with survivable types of OI, OI type III is most often de novo (85%), followed by type IV (50%) and type I (34%).[6]: Table 1
Some populations can have a higher incidence of OI than would be otherwise expected if they have a larger than average number of carriers of the recessive forms of the disease.[1]: 20–21 [141]
Other animals[edit]
In dogs, OI is an autosomal recessive condition, meaning that dogs with two copies of the allele will be affected.[157] Many breed organizations and veterinarians offer OI tests to tell if a dog is a carrier of OI.[157][158] To prevent OI, dogs who are heterozygous for OI should only be bred to non-carriers.[158]
Naturally occurring mutations causing OI have been found in Golden Retrievers, Dachshunds, and Beagles. OI has also been identified in zebrafish and mice.[159]
Although dogs, mice, fish, and humans are not genetically identical, some of these animal models have been officially recognized to represent the varying types of OI in humans. For example, homozygous oim/oim mice experience spontaneous bone fractures, small body size, and kyphosis, making them a model of OI type III. Meanwhile, heterozygous oim/+ mice appear normal but have bones which are quite a bit weaker than wild mice, making them a model for OI type I.[159][160] As in human OI, the location on the gene which is mutated affects the severity of resulting disease—the G859C Col1a1 mouse is a model for OI type II as affected mice all die in the perinatal period.[159]
Animal testing on identified animal models may lead to human therapies for OI.[159]